This is actually a synthesis I did on August 2024, so more than half a year ago. Dextromethorphan was used as starting material for this synthesis, for various reasons: a) my first attempt at using Phenanthrene as a starting material was "successful" but gave very bad yields (7 - 14%). I assume this might explain the popularity of the Grewe Cyclization for opioid syntheses in general, because using the easy, comfortable Phenanthrene route where you already have the fundamental nucleus that all morphinans have and only need to hydrogenate the B and C-ring while adding/substituting the necessary functional groups, seems to deliver very bad yields unfortunately. This ties in directly to my second reason which is b) the much more economic route of DXM -> Levorphanol conversion and c) DXM is not a controlled substance here in Germany. Any person here can import as much of it as they want as long as it is used for non-commercial purposes.
I bought 80g of DXM HBr from a pharmaceutical company specializing in APIs based in India. I will start my first opioid entry with Levorphanol since it is the most simple member of the morphinan family. Just as Morphine is the simplest 4,5-epoxymorphinan, so too is Levorphanol which is the reason why I specifically chose this opioid because I can use it later as starting material for designing more sophisticated and potent derivatives, similar to how hydromorphone is a derivative of morphine (the 7,8-hydrogenated ketone of morphine to be pedantic). I believe the specially concentrated binding affinity (not to be confused with analgetic potency) (-)3-hydroxy-N-morphinan has on the µ-OR as opposed to 4,5-epoxymorphinans could be key in developing opioids with an above average euphoric response. This is my main motivation for opioid research. To develop agonists with almost no sedative, analgesic attributes while hitting all the right buttons to trigger the euphoric response. You only need two of such agonists with no cross-tolerance to each other (like morphine and methadone) and boom: you have a medical protocol consisting of bi-weekly opioid rotation to indefinitely treat depression without loss of the good effects that comes with chronic use of cross-tolerant opioids. Such euphoria-only opioids could be marketed as next-gen antidepressants. Still addictive as hell (in that case perhaps even more addictive) but at least they work in actually making depressive patients feel good and motivated and have enough energy instead of pumping them full with these worthless and toxic tricyclical ADs they're selling.
Now let's get to the actual meat and bones of this post.
:::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::
Levorphanol Tartrate Dihydrate Profile
Name:
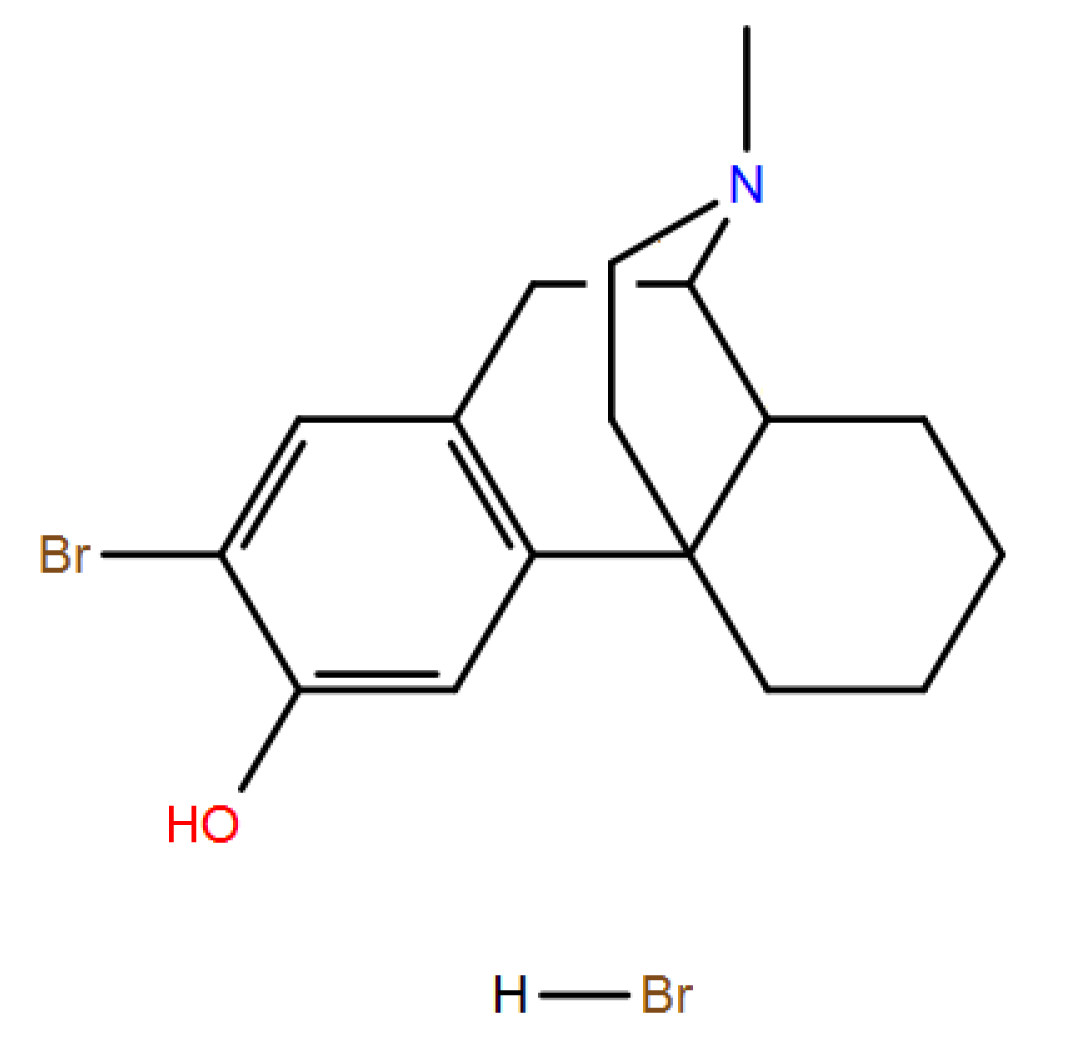
Levorphanol alias levo-3-hydroxy-N-methylmorphinan
Family:
Morphinan
INN:
Levorphanol, Aromarone, Dromoran
IUPAC name:
(1R,9R,10R)-17-Methyl-17-azatetracyclo[7.5.3.01,10.02,7]heptadeca-2(7),3,5-trien-4-ol
Other names:
(-)3-hydroxy-N-methylmorphinan
(-)17-methylmorphinan-3-ol
Stereoisomers:
Dextrorphan (+ isomer)
Racemorphan (+/- isomer containing both the opioid agonist Levorphanol as well as the dissociative drug Dextrorphan)
Crystallography:
Triclinic system
Salt forms tested incl. solubility ratio per unit water under normal conditions:
L. tartrate dihydrate (10.8), L. hydrochloride (7.9), L. hydrobromide (3.7); These ratios explain why the otherwise rarely seen tartrate dihydrate is the salt of choice for Levorphanol manufacture.
!!!WARNING!!!
Levorphanol is an opioid with a high degree of metabolic polymorphism, so the following figures can vary more or less tremendously from person to person.
Bioavailability:
PO: 70%
INS: ~70%
INH: 100% (tastes and smells like how I imagine mildly burned rice must taste like. Surprisingly non- chemical aroma.)
SC: 100%
IV: 100%
Potency:
PO: 8:1 (where 1 is morphine)
INS: 8:1
INH: 6:1
SC: 3.5:1
IV: 5:1
Onset of action:
PO: 0.25h
INS: 0.05h
INH: 15 sec
SC: 0.08h
IV: 9 sec
Peak response:
PO: 2 - 2.30h
INS: 1h
INH: 0.40h
SC: 0.45h
IV: 0.30h
Duration of action:
PO: 10h
INS: 6h
INH: 3.80h
SC: 4.20h
IV: 3h
Sedative response (1-10):
5.5
Euphoric response (1-10):
7
:::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::
Synthesis and Chemistry
Reagents:
- 3-methoxy-N-methylmorphinan hydrobromide
- Distilled H2O
- Any concentrated hydrohalic acid (50% HBr (aq) preferred, but 30% HCl (aq) is fine too)
- Any lower alcohol up to isopropanol (Methanol should work best)
- Any aprotic, non-polar solvent such as Trichloromethane
- Any strong amine base (30% NH3 (aq) works best)
- Tartaric acid
- Activated carbon
- Filter aid
The reaction mechanism itself couldn't be any simpler. The electrophilic methyl group of the aromatic ether is substituted with a good quality H+ entering group (provided by a hydrohalic acid) via a SEAR reaction, causing an electrophilic attack on the nucleophilic O- group and thereby replacing the methoxy group with an hydroxy group. This is achieved by mixing HBr (aq) (2.93 mol) with DXM HBr while heating it to reflux at 120°C for 150 minutes while stirring it with a magnetic stirring plate.
 |
Conversion from DXM HBr... |
 |
| ...to Levorphanol HBr |
This results in Levorphanol or (-)3-hydroxy-N-methylmorphinan hydrobromide as the main product with a methyl halogen as a byproduct (which is poorly soluble in water) and two other potentially hazardous byproducts, namely 10-ketolevorphanol and 2-chloro/bromolevorphanol. Halogenated pharmaceuticals are known to be neurotoxic and if HCl/HBr (aq) was used for the reaction, care must be taken to get the latter byproduct out of the opioid, preferrably down to ppm quantities, especially if injection is planned as chlorine atoms are small enough to bypass the BBB. The two byproducts were indeed detected and removed via a special recrystallization method (explained below).  |
Byproduct #1: 2-bromolevorphanol HBr
|
 |
Byproduct #2: 10-ketolevorphanol HBr
|
We will now move on to basify our Levorphanol HBr. After the above reaction is over, the mixture should be allowed to cool off to around 20°C and slowly added to a 2-phase mixture of NH3 (aq) (1.2 mol), distilled water (1.7 mol), methanol (0.69 mol) and chloroform (3.8 mol). Interestingly, methanol and chloroform shouldn't mix since the former is polar while the latter is a non-polar solvent, but that is just one more proof how theoretical chemistry often deviates strongly from practical reality.


After adding the NH3 (aq) to the TCM/Methanol mixture, two phases appear (the middle, milky white phase you see in the first photo only appeared temporarily after having poured the liquid ammonia into the system and then quickly disappeared for some reason. I have no idea what the hell that was and how that substance came to be): a top phase consisting of liquid ammonia and a bottom phase consisting of TCM/Methanol. Now stir the beaker for 15 minutes, put the mixture into a separating funnel and repeat the process for a total of 3x (obviously you throw away the aqueous phase, NOT the organic phase each time you're done).
 |
| Levorphanol freebase |
Notice how the organic phase is not transparent anymore. That's the Levorphanol base having been dissolved, creating a saturated solution (comparable to a saturated saline solution).
Now what's the purpose of this? Why not just basify the Levorphanol salt by letting it react with NH3 (aq) and filter the insoluble base out of the liquid? The problem is that the bromide anion and the hydrogen cation in the HBr salt bond with NH3 to form ammonium bromide (NH4Br). That is itself a salt which immediately undergoes a dissociation reaction in the aqueous phase, giving a positively charged NH4+ cation and a negatively charged Br- anion. The hydrogen proton of the NH4+ will then, by way of an AE reaction bond with the nucleophilic N of the Levorphanol base, making N electropositive as it donates an electron to the electrophilic proton. The hydrogen proton then bonds with the bromide anion and you end up with Levorphanol HBr again. All this happens too quickly for the Levorphanol base to dissolve in the organic layer. So trying to turn Levorphanol HBr into a base by letting it react with NH3 (aq) creates a never ending cycle of basification and neutralisation. To break this cycle we use an organic phase that employs solvents with the ability to dissolve the base only. The TCM/Methanol mixture does that. Unfortunately though, Br- anions are dissolvable in the organic phase too, which causes a certain portion of those anions to be present in the organic layer (16%). This means the bulk of the bromide gets extracted by NH3 (aq). Hence we make multiple extractions with fresh NH3 (aq), each time removing another 84% of the previous mass. The result is that the three extractions of the organic phase reduce the bromide impurity down to 0.41%. This is why I told you earlier to throw away the aqueous phase and not the organic one, because otherwise you'd be keeping the bromide anions while throwing away the delicious Levorphanol. Reminds me a little bit of some of those speedfreaks who follow the acetone wash guide on purifying their speed and accidentally end up throwing away the amphetamine while snorting the cut 😂
All right, now we have a Levorphanol base solution. Those who want to experiment with different salts can stop here and do the acid-base reaction with their acid of choice, while I'm going to turn the base into tartrate dihydrate. Anhydrous tartrate should be avoided as it tends to increase in hardness over time, thus making it difficult to chop and therefore sniff (kind of like acetone washed amphetamine sulfate that tends to take on this annoying shape of pellet-like powder where large quantities go right through your nose down into your throat as if you just snorted sand.
Take a separate beaker now and pour into it distilled water (0.5 mol) with an equal amount of tartaric acid. Heat it to 45°C and mix it well until everything has been dissolved. Maintain the temperature between 45 and 50°C during the whole process. Make sure the beaker with the base solution is stirring while you SLOWLY pour in the aqueous acid (increase temperature to 78°C). Just dumping the acid into the base all at once will cause the neutralization reaction to happen so incredibly fast that the solution will almost instantly solidify which will look like this:
This will then require of you to re-dissolve the Levorphanol Tartrate by pouring in more water which is not only kind of a messy process but will actually cause, for some reason that is unknown to me, great difficulty for the Levorphanol Tartrate to form crystals later on during the recrystallization procedure. You know you've done it correctly when no solids form during the reaction (see photos below).
.png) |
| Tartaric acid (aq) |
 |
| Levorphanol tartrate anhydrate |
In general, it is always a good policy to pour in liquid chemicals as slowly as possible into reaction mixtures. Inorganic chemistry might reward quickness, organic chemistry however will punish you for it. Keep stirring the L. tartrate solution for 15 minutes. The neutralization reaction above is slightly exothermic and will increase the temperature of your solution by approximately 3°C for a couple minutes.
What follows now is the recrystallization procedure mentioned earlier to isolate the potentially toxic byproducts 2-bromolevorphanol and 10-ketolevorphanol and end up with a truly pharmaceutical-grade purity profile. This is really the most important part of this entire synthesis, especially if you plan on shooting it. We know what Levorphanol does. We do NOT know what those other mystery substances do.
Now, after you've turned off the stirring magnet, slowly decrease the temperature down to room temperature over the course of a couple hours. The longer it takes for the temperature to fall, the bigger and more beautiful the crystal formations will be, regardless of the crystalline compound in question. That is almost a law of nature. Once the solution has reached room temperature, put the beaker into your refrigerator. Make sure to cause the least amount of movement to the solution as possible while walking to the refrigerator. Allow the beaker to remain in there until no more crystals form. This can take a couple of hours, an entire night or even days, depending on too many factors to count here. Once you no longer see more crystals forming, put the beaker into the freezer. Same principle of waiting applies here.







 |
See the orange teint of the crystals? Those are the 2-bromo impurities.
Visible thanks to the bromine atoms that cause orange discoloration.
You don't have that luxury with oxygen atoms which is why you don't
see the 10-keto impurities even though they are still there! |
What you see in the pictures above is L. tartrate anhydrate. Pulverize the crystals, put them in your oven on 60°C and wait for 4 - 6 hours. It really needs to be dirt dry because even a little bit of humidity will cause the product yield to suffer. The remaining solvent mixture in your beaker contains a large portion of the byproducts, which you can either throw away or you can distill the solvents out and use them for another project if you are a poor chemist (actually, it's more expensive to win back the solvents through distillation due to electricity costs, rather than simply buying a new bottle of TCM, Methanol, etc., but I suppose that depends on the quantity and where you live. Energy costs here in Germany have become a real nightmare.). Clean your beaker or take a new one and fill it with distilled water (2.4 mol), put activated carbon (0.024 mol) and filter aid (0.012 mol) into it, suspend the dry L. tartrate anhydrate powder into the mix, heat it to 84°C and stir it for 60 minutes. Vacuum filter the mixture into a flask and let it cool slowly as described above. What you'll get now are L. tartrate dihydrate crystals. Dry it again, prepare a 90% aqueous methanol solution (or any aqueous, lower alcohol above 80%), dissolve the dried L. tartrate dihydrate in it, heat it to 84°C again, stir for 60 minutes and let it cool off. You'll get the anhydrous salt again. These previous two recrystallizations essentially purified the Levorphanol from the two byproducts mentioned above. If you take opioids only perorally you can dry your L. tartrate anhydrate and call yourself the proud owner of 99.63% pure Levorphanol and proceed to get high as a kite. I however will recrystallize the salt back to its dihydrous form because I prefer to sniff and occasionally shoot. Repeat the process above only without the activated carbon and the filter aid and you'll get L. tartrate dihydrate.
Note: I forgot to photograph the latter two recrystallizations. What essentially happened was the orange teint disappeared with every recrystallization until the crystals became as white as a snowflake. Note to self: don't do lab work while high.
 |
| The L. tartrate dihydrate crystallized around my damn thermometer 😂😂😂 |

 |
I just couldn't resist and had to press some of the crystals 😊
Imagine if I gave this to people telling them it was "fishscale cocaine". They'd
be dying left and right 💀
Btw, this is still somewhat wet because dihydrous L. tartrate is practically
impossible to press. It's just too fluffy to hold itself together like that. |
 |
And now it looks like crack. Imagine if I gave this to people telli...ah you know how it goes.
Completely dried. Look at those BEAUTIFUL crystals 😍😍😍 |
 |
Now isn't that a nice yield? 91% of the product remained. In other words, 9% lost to
impurities and mass entropy. Couldn't be happier. |
 |
| This is keeping me totally chilled indeed 😁😁😁 |
 |
| Levorphanol Tartrate Dihydrate |













.png)












